Practical information
Pangenome file
PPanGGOLiN operates by creating and managing pangenomes within a specialized file format known as HDF-5 (identified by the .h5 extension in the result directory). This unique file serves as a central repository, recording the outcomes of pangenome analyses along with the associated parameters and complete pangenomic content.
The benefit of this approach is the ability to exclusively store and manipulate pangenomes using PPanGGOLiN through this single file. It encapsulates all pertinent information regarding the analysis performed, including parameters applied and the entirety of the pangenomic data. It can be used to generate or regenerate flat output files describing the pangenomes content with dedicated commands (write_pangenome, write_genomes, fasta and draw).
Once the pangenome is established, including gene clustering, partitioning, optional prediction of Regions of Genome Plasticity (RGP), Spots of insertions, and functional modules, it becomes a versatile input for various PPanGGOLiN commands. For instance, the msa command generates multiple sequence alignments of genes, the projection command annotates external genomes with the pangenome content, and the context command identifies genomic contexts of proteins of interest.
PPanGGOLiN utilizes the pytable Python library for handling the pangenome HDF-5 file. While graphical user interface tools like ‘ViTables’ allow manual exploration and editing of the file, it’s cautioned against, as it may compromise compatibility with PPanGGOLiN.
You can use the command info to get comprehensive insights into the contents and construction process of a pangenome file.
ppanggolin info -p pangenome.h5
Required computing resources
Most of PPanGGOLiN’s commands should be run with as many CPUs as you can give them by using the –cpu option as PPanGGOLiN’s speed increases relatively well with the number of CPUs. While the ‘smallest’ pangenomes (up to a few hundred genomes) can be easily analyzed on a normal desktop computer, the biggest ones will require a good amount of RAM. For example, 40 strains of E. coli were analyzed in 3 minutes using 1.2Go of RAM using 16 threads. 1000 strains were analyzed in 45 minutes with 14 Go of RAM using 16 threads, and as of writing those lines (October 2019), 20 656 genomes was the biggest pangenome we did, and it required about a day and 120 Go of RAM. The following graphic can give you an idea of the time it takes for a pangenome analysis given the number of genomes in input.
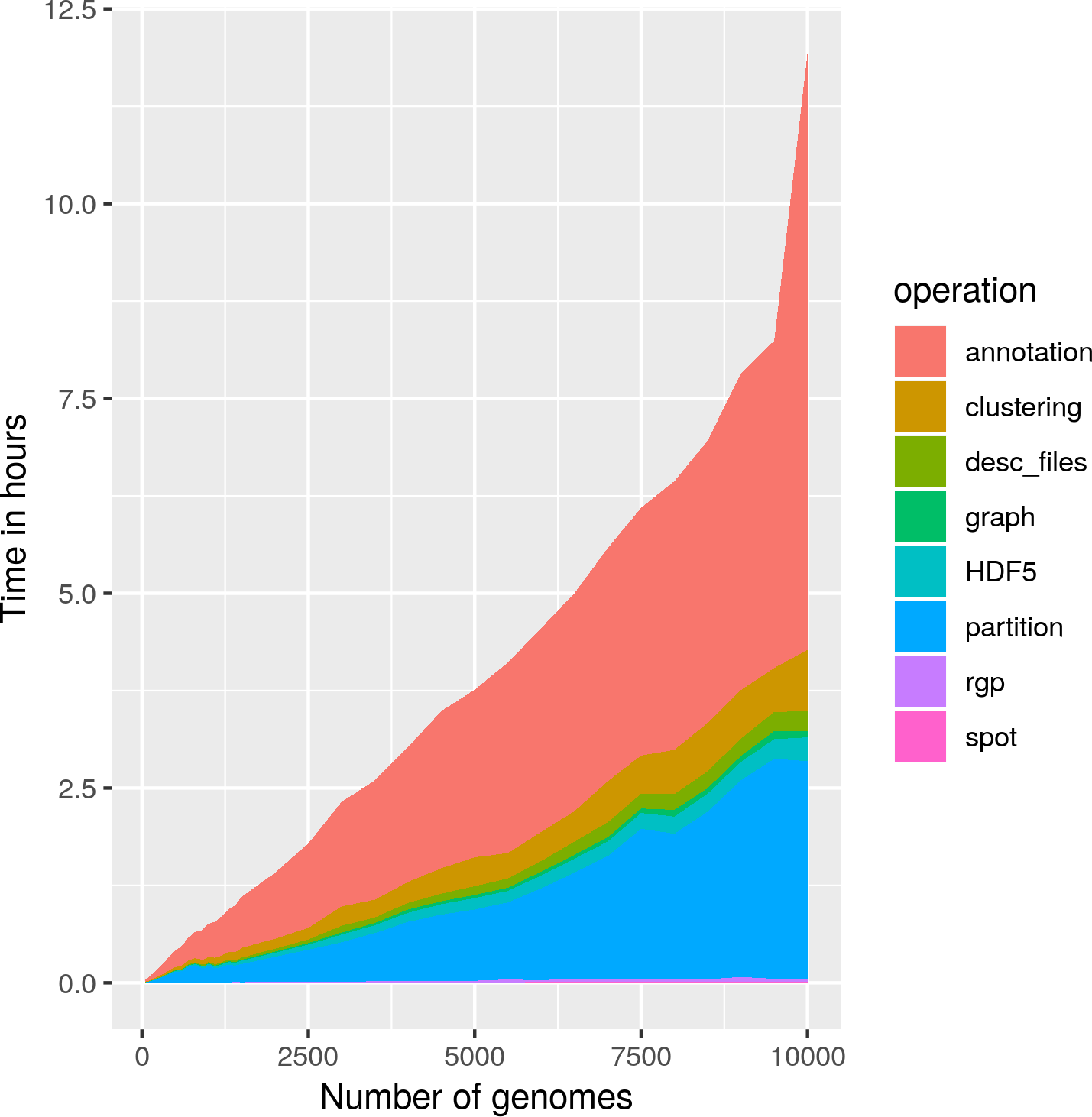
This data was collected using PPanGGOLiN v1.1.72.
Usage and basic options
As most programs in bioinformatics, you can always specify some utility options.
You can specify the number of CPUs to use (which is recommended ! The default is to use just one) using the option --cpu.
You can specify the output directory (if not provided, one with a unique random name will be generated) using the option --output.
If you work in a strange environment that has no, or little available disk space in the ‘/tmp’ (or your system equivalent, what is stored in TMPDIR) directory, you can specify a new temporary directory using --tmp
If you want to redo an analysis from scratch and store it in a directory that already exists, you will have to use the --force option.
Be wary, however, that the data in that directory will be overwritten if named identically as any output file written by ppanggolin.
PPanGGOLiN is deliberately very verbose, to help users understand each stage of the analysis.
If you want, verbosity can be reduced in several ways.
First, you can specify the verbosity level with the --verbose option.
With 0 will show only warnings and errors, 1 will add the information (default value), and if you encounter any problem you can use the debug level with value 2.
Then you can also remove the progress bars with the option --disable_prog_bar
Finally, you can also save PPanGGOLiN logs in a file by indicating its path with the option --log.
Configuration file
Advanced users can provide a configuration file containing any or all parameters to PPanGGolin commands.
This feature is particularly useful for workflow commands such as workflow, all, panrgp, and panmodule, as it allows for the specification of all parameters for each subcommand launched in a workflow.
Additionally, a configuration file can be used to reuse a specific set of parameters across multiple pangenomes.
To provide a configuration file to a PPanGGolin command, use the --config parameter.
Note
Any command line arguments provided along with a configuration file will override the corresponding arguments specified in the configuration file. When an argument is not specified in either the command line or the configuration file, the default value is used.
The configuration file is a JSON file that contains two sections common to all commands: input_parameters and general_parameters.
In addition, there is a section for each subcommand that contains its specific parameters.
You can generate a configuration file template with default values by using the ppanggolin utils command as follows:
ppanggolin utils --default_config CMD
For example, to generate a configuration file for the panrgp command with default values, use the command
ppanggolin utils --default_config panrgp
This command will create the following configuration file:
input_parameters:
# A tab-separated file listing the genome names, and the fasta filepath of its
# genomic sequence(s) (the fastas can be compressed with gzip). One line per genome.
# fasta: <fasta file>
# A tab-separated file listing the genome names, and the gff/gbff filepath of
# its annotations (the files can be compressed with gzip). One line
# per genome. If this is provided, those annotations will be used.
# anno: <anno file>
general_parameters:
# Output directory
output: ppanggolin_output_DATE2023-04-14_HOUR10.09.27_PID14968
# basename for the output file
basename: pangenome
# directory for storing temporary files
tmpdir: /tmp
# Indicate verbose level (0 for warning and errors only, 1 for info, 2 for debug)
# Choices: 0, 1, 2
verbose: 1
# log output file
log: stdout
# disables the progress bars
disable_prog_bar: False
# Force writing in output directory and in pangenome output file.
force: False
annotate:
# Use to not remove genes overlapping with RNA features.
allow_overlap: False
# Use to avoid annotating RNA features.
norna: False
# Kingdom to which the prokaryota belongs to, to know which models to use for rRNA annotation.
# Choices: bacteria, archaea
kingdom: bacteria
# Translation table (genetic code) to use.
translation_table: 11
# In the context of provided annotation, use this option to read pseudogenes. (Default behavior is to ignore them)
use_pseudo: False
# Allow to force the prodigal procedure. If nothing given, PPanGGOLiN will decide in function of contig length
# Choices: single, meta
prodigal_procedure: False
# Number of available cpus
cpu: 1
Issues, Questions, Remarks
If you have any question or issue with installing, using or understanding PPanGGOLiN, please do not hesitate to post an issue in github! We cannot correct bugs if we do not know about them, and will try to help you the best we can.
Before reporting a bug, if possible please add the option --verbose 2 to your command to provide additional information. Please also add some context and tell us what you were trying to do.
Citation
If you use this tool for your research, please cite:
Gautreau G et al. (2020) PPanGGOLiN: Depicting microbial diversity via a partitioned pangenome graph. PLOS Computational Biology 16(3): e1007732. https://doi.org/10.1371/journal.pcbi.1007732
If you use this tool to study genomic islands, please cite:
Bazin et al., panRGP: a pangenome-based method to predict genomic islands and explore their diversity, Bioinformatics, Volume 36, Issue Supplement_2, December 2020, Pages i651–i658, https://doi.org/10.1093/bioinformatics/btaa792
If you use this tool to study modules, please cite:
Bazin et al., panModule: detecting conserved modules in the variable regions of a pangenome graph. biorxiv. https://doi.org/10.1101/2021.12.06.471380